













Advances in genomics has led to the discovery of multiple predisposition genes linked to increased risk for gastrointestinal (GI) cancer. The goal of this review is to assist physicians and allied health care professionals in understanding the current paradigm shift in clinical genetic testing for hereditary GI cancer predisposition syndromes; with focus on multigene panel testing (MGPT) and test results interpretation. Additionally, this review introduces direct-to-consumer and at-home genetic testing. Both delivery models are increasing in popularity and clinicians will be expected to address results from patients who utilize these approaches.
Recent Findings:
Technological advancement and reduced costs have transformed the genetic testing approach from single syndrome genetic testing to broad-based MGPT. MGPT has the benefit of aiding in efficient genetic diagnosis; however, clinicians should be knowledgeable of possible results including variants of uncertain significance, secondary findings, and pathogenic variants within high- and low-moderate-risk genes, as well as genes for which risks are ill-defined.
Summary:
The landscape of clinical cancer genetics continues to evolve rapidly. Timely updates are critical to ensure the medical community is familiar with current considerations and ongoing challenges regarding genetic testing for hereditary GI cancer susceptibility.
Keywords: Multigene panel testing, variant interpretation, gastrointestinal cancer susceptibility, emerging, direct-to-consumer, genetics
Go to:
Introduction
Over the last several decades, advances in genomics have led to the discovery of multiple predisposition genes linked to increased risk for gastrointestinal (GI) cancer (1). While several predisposition syndromes are well-characterized with available consensus and/or evidence-based management protocols, others are newly appreciated and/or emerging with less clear clinical guidelines (1). As genetic testing expands, results interpretation has become more complex and nuanced. The goal of this review is to assist physicians and allied health care professionals in understanding the current paradigm shift in clinical genetic testing for hereditary gastrointestinal cancer predisposition syndromes.
Gastrointestinal cancer risk genes identified through genetic testing
There is a continually growing number of GI cancer risk genes that can be identified through clinical genetic testing (1, 2). Germline hereditary cancer testing can include analysis of genes that are related to high-risk cancer syndromes, low-to-moderate risk cancer susceptibility, or may include genes with emerging-evidence for cancer susceptibility.
Traditionally, high-risk colorectal cancer (CRC) risk genes are categorized related to the clinical presentation of “polyposis” or “nonpolyposis.” Well-defined, high-risk, colonic adenomatous polyposis conditions include familial adenomatous polyposis (FAP), attenuated FAP (AFAP) and MUTYH-associated polyposis (MAP) [Table 1]. Additionally, classically defined hamartomatous polyposis conditions include juvenile polyposis syndrome (JPS), Peutz-Jeghers syndrome (PJS) and PTEN-hamartoma tumor syndrome, sometimes referred to as “Cowden Syndrome” (CS). There are also high-risk syndromes without polyposis that increase CRC risk such as Lynch syndrome (LS) and Li-Fraumeni syndrome (LFS). Furthermore, there are other high-risk cancer syndromes that increase risk for extracolonic GI cancers. For example, hereditary diffuse gastric cancer syndrome (HGDC) is caused by “likely pathogenic or pathogenic” (LP/P) variants [Figure 1] in the CDH1 gene and significantly increases risk for diffuse gastric cancer (3). Overall, the risk of developing GI related cancers is significantly higher than the general population in the setting of a high-risk syndrome; exemplified by the 50-80% lifetime risk of developing CRC in some individuals with LS compared to the general population risk of 5% (4–6).
Table 1:
In addition to the high-risk hereditary GI cancer syndromes outlined above, there are specific variants and/or genes that are considered low-to-moderate risk [Table 1]. Generally speaking, LP/P variants in moderate risk genes can confer a roughly 2-fold increased risk in CRC risk, which is a similar level of risk as having a first degree relative with CRC (7, 8). One common moderate risk variant is p.I1307K in the APC gene, frequently referred to as APC*I1307K. This variant is present in 7% of individuals with Ashkenazi Jewish ancestry (9). Importantly, individuals with this variant do not have FAP or AFAP, but have been shown to have an elevated risk of CRC (9, 10). An example of a low risk variant is a monoallelic LP/P variant in the MUTYH gene (“monoallelic” defined as an individual with a single MUTYH LP/P variant). Monoallelic MUTYH LP/P variants are present in ~2% of the general population (11). Individuals with a monoalleic MUTYH LP/P variant differ drastically from those with biallelic MUTYH LP/P variants who have the highly penetrant autosomal recessive condition, MAP. The CRC risk estimates for monoallelic MUTYH LP/P variants are conflicting, and there is no consensus about whether these individuals have increased CRC risk (7, 12). Given that many of these low-to-moderate risk genes have conflicting risk estimates, the cancer screening recommendations can be controversial, and will likely evolve over time (7).
A third class of genes potentially identified on genetic testing are those with emerging evidence to support possible cancer risk [Table 1]. Many of these genes currently have limited information regarding level of cancer risk and cancer spectrum, often making it challenging to definitively associate cancer risk. Genes with emerging evidence also do not typically have any evidence-based guidelines for medical management, however some may have guidelines based on expert opinion (13). Further details about these emerging-evidence genes are beyond the scope of this current review.
Clinical Genetic Testing
Historically, based upon the satisfaction of certain clinical criteria, a patient was offered genetic testing for a particular hereditary cancer syndrome. This testing typically focused on a single hereditary cancer syndrome, with limited evaluation of one or a few genes. If such testing was positive, the LP/P variant would have established associations with the high-risk syndrome of interest, with a resulting significantly elevated cancer risk without intervention. Traditionally, families were referred to genetics programs staffed by genetic counselors and housed within academic institutions, and they were ascertained and referred based upon a reasonable a priori or pre-test probability of testing positive for a disease-associated variant. Genetic testing was guided by clinical suspicion and was not typically offered (with explanation) if a priori chance was insufficient, as single high-risk cancer predisposition syndromes remain relatively rare in the general population. Technological advancement and reduced costs have transformed the genetic testing approach from single syndrome genetic testing to more broad-based multigene panel testing (MGPT) (14, 15). MGPTs may evaluate some or all known cancer predisposition syndromes simultaneously regardless of a patient’s presenting clinical phenotype or family history. Commercial diagnostic laboratory (CDL) test offerings range from one to greater than eighty genes on a single panel, or test. CDLs may also include preliminary or emerging-evidence genes that have limited data regarding disease association and unknown/unstudied clinical utility. Clinical gene panels can be categorized as the following:
1.Cancer syndrome-specific*
2.Cancer-site specific panel*
3.Pan-cancer panel*
Single gene panel incorporating hereditary cancer risks across multiple organ systems +/− emerging evidence genes (i.e. colon, pancreatic, prostate, breast, ovarian, endocrine, etc.)
Several CDLs offer panel customization
Correct ordering and interpretation of genetic test results is an existent problem in the medical community, and predates the integration of MGPT (16). By its very nature, MGPT has the capability of returning more information than single-gene testing. MGPT is heavily utilized in the clinical genetics community; however, at present, there is no standard of care regarding when to offer MGPT. Given the lack of expert consensus or guideline regarding the application of MGPT, it is important that one can recognize the benefits, as well as risks and limitations of MGPT [Table 2].
Table 2:

At-home Genetic Testing At-home (AH) genetic testing is the opportunity for a patient/consumer to conduct clinical genetic testing, such as MGPT, outside the traditional consultative medical model. It is discussed as part of this review given their growing popularity and the increasing likelihood that a practitioner will be presented with or asked to facilitate AH genetic testing. Procedurally, a patient requests genetic testing and a test kit is mailed to the patient’s home, which is approved online by their own physician or a physician employed by the testing company. AH genetic tests are clinical diagnostic tests and national professional societies such as the National Society of Genetic Counselors (NSGC) recognize their potential to expand access to genetic testing (17). However, in order for AH genetic testing to be conducted responsibly, only clinical-grade testing interpreted by healthcare professionals with genetics expertise should be used to inform decisions about preventive care, diagnosis, or medical management. Furthermore, it is important to know the laboratory accreditations, as well as test accuracy and reliability (17).
Direct-to-Consumer Genetic Testing
Direct-to-Consumer (DTC) genetic tests are widely advertised and sold directly to the public. DTC genetic testing does not require physician involvement as such tests are not clinically diagnostic nor intended for medical use (18). DTC genetic testing is included as part of this review because such companies and tests are prevalent and it is important to know their benefits as well as distinct risks and limitations. DTC tests offer information that may include ancestry, carrier status (such as cystic fibrosis or sickle-cell disease), predicted drug response, and non-disease traits such as eye color. While DTC tests increase access and may provide interesting health and ancestry insights, these tests are not ordered through a healthcare provider, are not diagnostic, and offer risk information for only a limited set of conditions (18). More so, DTC testing utilizes different technology and protocols compared to clinical genetic testing. Taken together, DTC has the potential to create misunderstandings regarding cancer risk as illustrated below.
Hypothetical scenario: An unaffected individual desires a genetic evaluation for Lynch syndrome based upon a family history of CRC developing prior to age 50 in her mother as well as several maternal aunts and uncles. This individual orders a “health profde screen ” through a DTC company that was advertised on social media. The individual ordered their test online, received the kit at their home, provided saliva in a tube, and mailed the kit back to the testing lab. The results, released through the company’s online portal, reveal an average to modest elevation in CRC risk. The individual believes the evaluation for inherited CRC risk was complete and comprehensive. Furthermore, she interprets her risk to develop CRC as average-modest despite her strong family history.
Explanation: Most DTC companies evaluate single nucleotide polymorphisms (SNPs) across the genome. By definition, a SNP is a genomic variant, common in the population, with small contribution to disease risk (relative risk close to 1). In combination, multiple SNPs may predispose to disease development; however, SNP-based analysis does not include the evaluation of highly penetrant genes linked with a dominant pattern of cancer. In context, for our individual, comprehensive risk assessment for Lynch syndrome was not completed. This DTC test did not evaluate, via full sequencing and copy number analysis, for LP/P variants within MLH1, MSH2, MSH6, PMS, and EPCAM, and it remains possible the individual does carry a Lynch-associated variant.
As illustrated here, it is important to be aware that DTC tests are not clinically validated for medical use and do not provide a comprehensive risk assessment. Another recent concern developing with DTC testing is that some testing companies provide customers their raw genotyping data upon request, which may include variants in genes linked to GI cancer predisposition syndromes. While DTC companies do not provide interpretation of the raw data and include disclaimers regarding lack of validation, there are third party companies that offer interpretation for a fee (19). In one particular study by a “Clinical Laboratory Improvement Amendments-College of American Pathologists” (CLIA-CAP) designated commercial diagnostic laboratory, 49 variants previously identified by DTC testing were sent for clinical confirmatory testing, and 40% were not confirmed, and thus deemed false positives. Additionally, eight variants designated as “increased risk” in the DTC raw data file or by 3rd-party interpretation, were classified as “benign” by the CLIA-CAP commercial diagnostic laboratory (18). Due to concerns such as these, per the NSGC, any genetic variants identified in raw data files should be confirmed in a clinical laboratory prior to being using for any medical decision making (17).
Classification of genetic testing results
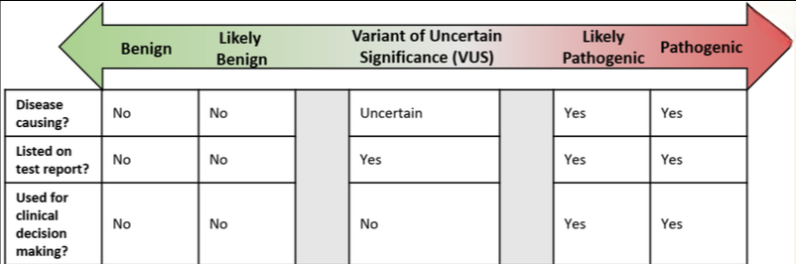
Genetic testing results can be placed in three separate categories: Positive, negative and variant of uncertain significance [Figure 1]. Positive genetic testing results consist of the identification of a LP/P variant. Negative genetic testing results are unremarkable or may have identified likely benign or benign (LB/B) variants that are not standardly listed on the genetic testing report. The American College of Medical Genetics and Genomics (ACMG) recommends the use of standard terminology to classify variants identified in genes that are associated with single gene disorders (20). This terminology is described below and summarized in Figure 1.
Pathogenic: A clinically significant variant is detected with strong evidence to support it is disease causing. If the condition is inherited in an autosomal recessive manner, a single pathogenic variant may not be sufficient to cause disease by itself (i.e. monoallelic MUTYH variant). Healthcare providers can make medical management recommendations based on this information. Additionally, family members can pursue targeted testing for the pathogenic variant.
Likely Pathogenic: A clinically significant variant is detected with evidence to support with greater than 90% certainty the variant is disease causing, although there is not enough evidence to prove pathogenicity conclusively. Healthcare providers can make medical management recommendations based on this information. Additionally, family members can pursue targeted testing for the likely pathogenic variant.
Variant of Uncertain Significance (VUS): There is limited and/or conflicting information regarding the variant; the variant does not have enough evidence to support the categorization of “LP/P” nor “LB/B”. In the majority of cases healthcare providers should not use VUS to make medical management recommendations, and should instead base their recommendations on personal and family history.
Likely Benign: A variant which is not likely to be clinically significant is detected with evidence to support with greater than 90% certainty that it is benign (i.e. not disease causing). Healthcare providers should make medical management recommendations based on personal and family history.
Benign: A variant which is not clinically significant is detected with strong evidence that it is benign (i.e. not disease causing). Healthcare providers should make medical management recommendations based on personal and family history.
The ACMG, CAP, and the Association for Molecular Pathology (AMP) have proposed standards and guidelines for laboratories to interpret variants within analyzed genes (20). Variant curation requires integrating multiple pieces of information to help determine pathogenicity, such as variant frequency in the population, conservation data, computational tools, functional studies, and clinical features. Despite attempts to help create a more uniform variant classification process, there are still discrepancies in classification among different reputable genetic laboratories (21). A study evaluating data from a self-enrolled online genetic registry called Prospective Registry of Multiplex Testing (PROMPT), reported that 11% of participants had a variant with conflicting interpretation that would have a clinically significant impact (i.e. some labs reported LP/P while other labs reported VUS) (22). This type of discordant result may alter medical management for the individual in addition to having implications for family members. Many laboratories participate in data sharing and submit variants along with their interpretations to ClinVar, which is supported by the National Center for Biotechnology Information, part of the National Institutes of Health (23). ClinVar (https://www.clinvar.com) is a free public resource that organizes specific data regarding variants, which allows users easy comparison of variant interpretations across different participating laboratories.
Medical decision making based on genetic testing results
Likely Pathogenic/ Pathogenic (Positive) When a pathogenic or likely pathogenic variant appears on a genetic test report, it is considered a “positive” result. A positive genetic test should be put in context of the individual’s personal and family history to determine whether it provides a relevant genetic explanation.
For individuals with a personal history of cancer consistent with the genetic finding, it may help confirm the diagnosis of a hereditary cancer syndrome. In other words, the LP/P variant is identified in a gene with an established association with the reported phenotype (i.e. clinical presentation). Finding out this information may help identify if the affected individual has an elevated risk for developing another cancer which may lead to interventions to help reduce risk. Additionally, it may help guide optimal treatment for the individual’s current malignancy. For individuals without a personal history of cancer who have a family history consistent with the genetic finding, the positive result would be considered “predictive”, and confirmatory for increased cancer risk.
It is also possible to identify an “incidental” genetic finding when MGPT is performed. An incidental or secondary finding is when an individual receives a positive result in a gene that was un-anticipated based on the personal/family history. Secondary findings are most likely to consist of low-to-moderate risk gene variants (i.e. APC*I1307K and monoallelic MUTYH) since many of the low-to-moderate risk genes are observed at a relatively high frequency in the general population (9, 11). Caution should be exercised when interpreting these low-to-moderate risk secondary findings; it is important to prevent assigning causality or over-associating these findings with the personal and family history. In most cases, these findings are incidental and are not associated with the clinical indication for testing [Table 3].
Secondary, or incidental, findings can also be observed with genes associated with a high-risk cancer syndrome. In scenarios when an individual is found to carry a mutation in a high-risk cancer gene that does not align with the personal/family history; clinical recommendations and cancer risk estimates may be more challenging:
Hypothetical scenario: An individual with a family history of pancreatic cancer and no family history of gastric or breast cancer undergoes MGPT which identifies a LP variant in the CDH1 gene. This incidental finding may not explain the family history of pancreatic cancer for which the testing was originally ordered. However, once an incidental finding is identified an additional challenge becomes management of this unexpected finding in the absence of a personal or family history that is consistent with the expected syndrome phenotype. Healthcare providers specializing in hereditary cancer genetics can help navigate these scenarios in which the genetic testing result appears to be discordant with the personal and/or family history.
Individuals with a positive genetic testing result are strongly encouraged to share their genetic results with other biological family members. Testing other family members, termed “cascade testing”, plays an important role in identifying other at-risk individuals who may benefit from earlier and more frequent surveillance and/or risk-reducing surgeries. Despite the importance of testing relatives, cascade testing is significantly underutilized among families (24). Some of the most commonly cited barriers for cascade testing included limited understanding or knowledge among the individual, relative or healthcare provider in addition to poor communications skills (25). As healthcare providers, it is important to acknowledge the implications for family members and to help encourage dissemination of genetic information.
Cascade testing is especially pertinent for high-risk cancer syndromes such as Lynch syndrome, the most common inherited CRC syndrome (26). Those identified to harbor a mutation in any one of the Lynch syndrome genes are recommended to undergo intensive surveillance including colonoscopy starting between age 20-25; an empirically proven strategy to reduce mortality (27). In addition to identifying relatives at a higher risk of cancer, cascade testing also has the ability to determine who tests negative for the familial LP/P variant; otherwise known as a “true negative” result.
Predictive testing for familial low-to-moderate risk variants may not be as impactful in certain individuals. In some scenarios, the magnitude of risk associated with a positive result may not warrant increased surveillance (or preventive surgery) beyond that justified by family history alone (28). These nuanced conversations are best navigated by healthcare providers specializing in cancer genetics.
There are also reproductive considerations for individuals who have a positive genetic test result. Couples have the option to consider use of reproductive technologies, specifically PGT-M (preimplantation genetic testing for monogenic/single gene defects) in combination with in vitro fertilization. PGT-M significantly reduces the chance of passing on a known LP/P variant to the next generation. Those who are interested in PGT-M may discuss in greater detail with their genetics providers and may be referred to a fertility clinic specializing in this service for more information.
Variant of uncertain significance Variants of uncertain significance (VUS) are increasingly identified as genetic testing becomes more common and multigene panels include more genes. If an individual is tested using a syndrome specific panel for the five Lynch syndrome genes, the VUS rate is about 5% (29). However if a larger pan-cancer panel is performed, the VUS frequency increases to 31-40% (30). Additionally, VUS are more commonly encountered in non-Caucasian populations, which is in part related to lower testing volume and less genetic data from ethnically diverse populations (31).
VUS may be reclassified over time once more evidence becomes available. The reclassification process does not require re-testing the original sample, but does require the laboratory to obtain more information about whether the specific variant impacts the gene function or has associated clinical features. Approximately 90% of the time when a VUS is reclassified, it is downgraded to “likely benign” or “benign” (32). However, it is possible for a VUS to be upgraded to “likely pathogenic” or “pathogenic”, which would then likely have significant implications for medical management and familial cascade testing. Given the potential for a VUS to be reclassified, it is important for individuals with a VUS to maintain contact and follow-up with a genetic provider in the event new information is made available.
Medical management should not be altered based on a VUS given the lack of certainty about the meaning of the result (20). Additionally, testing family members for the VUS is not recommended given that it will not impact clinical decision-making. Consultation with a cancer genetic specialist may help interpret the VUS in context of clinical features and establish whether additional surveillance may be indicated based on the personal and/or family history. There are rare exceptions where it may be deemed appropriate for a VUS to be used clinically due to discordant variant interpretation; this should only be considered after consulting a healthcare professional with an expertise in hereditary cancer genetics.
Negative A negative genetic test result indicates that the laboratory did not identify a reportable variant. Most laboratories do not routinely include LB/B variants on reports given that those variants are not associated with disease. Interpretation of a negative genetic test result can be complicated and is based on the context of the individual’s clinical features and family history. Genetic counselors are experts at integrating this information to interpret a negative result and determine the next best steps.
Examples of how interpretation of a negative genetic testing result can become complicated:
When there is a high a priori chance for an individual to test positive, a negative genetic testing result cannot exclude the possibility of hereditary cancer risk. It is possible additional genes not tested (or presently known) explain the personal/family history. It is also possible a LP/P variant in one of the genes evaluated is not detectable by current genetic testing technology. Specific types of gene variants such as structural rearrangements (inversions, translocations, etc.) or deep intronic variants that may impact splicing are not typically detectable using most current methodologies (33).
If the person tested is unaffected with cancer, a negative genetic test result is considered “uninformative”. In this scenario, the reason for the family history of cancer remains unclear. The family history of cancer could be due to reasons unrelated to the genes evaluated or it is possible there is a detectable LP/P variant in one of the genes analyzed in other family members. When possible, it is ideal to test affected family members with cancer to best inform risk in an individual or family.
Introduced previously, one of the most informative negative genetic testing results is the “true negative.” A “true negative” applies to individuals who are negative for a known familial LP/P variant in a high-risk hereditary cancer syndrome. If an individual is a “true negative”, they did not inherit the determined source of cancer risk in their family and thus, their cancer risk remains similar to the general population. For example, a child is born to a parent with an established pathogenic variant in the APC gene associated with FAP. Unlike many adult onset hereditary cancer syndromes, FAP has pediatric manifestations and therefore it is appropriate to perform predictive testing in a minor given the implications for medical management (34). Evaluation of the child for the familial APC variant is negative, with the pathogenic variant not detected. This indicates the child tested is a “true negative,” which is a significant result as it confirms the child does not have FAP, and prevents unnecessary invasive surveillance starting at a young age (13). Alternatively, if the child never had testing, guidelines would recommend colonoscopy starting around age 10-12 based on a family history of FAP (13).
There are some caveats in interpreting “true negative” results. A full family history should be collected and evaluated by a genetic counselor or a physician that specializes in hereditary cancer to ensure there is no remaining family history of cancer that may not be explained by the known high-risk gene LP/P variant. Additionally, there is a lack of data to support the application of this concept to low-to-moderate risk genes. At this time, testing negative for a known familial low-to-moderate risk LP/P variant may not justify reduction of surveillance and does not provide the same clarity when compared to testing negative for a familial high-risk gene LP/P variant (28).
Go to:
Conclusion
The traditional approach to gastrointestinal hereditary cancer risk assessment has transitioned from phenotype-driven genetic testing to MGPT. MGPT has the advantage of detecting germline LP/P variants that would have potentially remained undiscovered based upon phenotype and clinical guidelines alone (35). Although MGPT has many advantages, there are also challenges: variant interpretation, unexpected findings, and the management of carriers of LP/P variants for which the optimal management has not been clearly elucidated (35). Additionally, clinicians need to be familiar with the differences between clinical laboratories, DTC testing and test options as accessibility to genetic and genomic testing increases. It is likely that patients will bring genetic testing reports with them to their appointments with the expectation of guidance in results interpretation and integration into their own medical care. Ultimately, it is not the expectation that the physician or allied health professional be an expert in genetic testing but that there is reasonable proficiency in understanding what a result might mean (or not mean) and when to seek counsel from the laboratory directly, or refer to specialized cancer genetic services (https://www.fmdageneticcounselor.com/). The landscape of clinical cancer genetics continues to evolve rapidly. Timely updates are critical to ensure the gastroenterology as well as the medical community as a whole is familiar with current considerations and ongoing challenges regarding genetic testing for hereditary GI cancer susceptibility.